
Решение некоторых из основных задач 21-го века, таких как производство чистого электричества или разработка высокотемпературных сверхпроводников, потребует от нас разработки новых материалов с особыми свойствами. Чтобы сделать это на компьютере, требуется моделирование электронов, субатомных частиц, которые управляют тем, как атомы соединяются в молекулы, а также ответственны за поток электричества в твердых телах. Несмотря на десятилетия усилий и несколько значительных достижений, точное моделирование квантово-механического поведения электронов остается открытой задачей. Теперь, в бумага (PDF с открытым доступом), опубликованном в журнале Science, мы предлагаем DM21, нейронную сеть, достигающую современной точности в больших областях химии. Чтобы ускорить научный прогресс, мы также открываем исходный код код для любого использования.
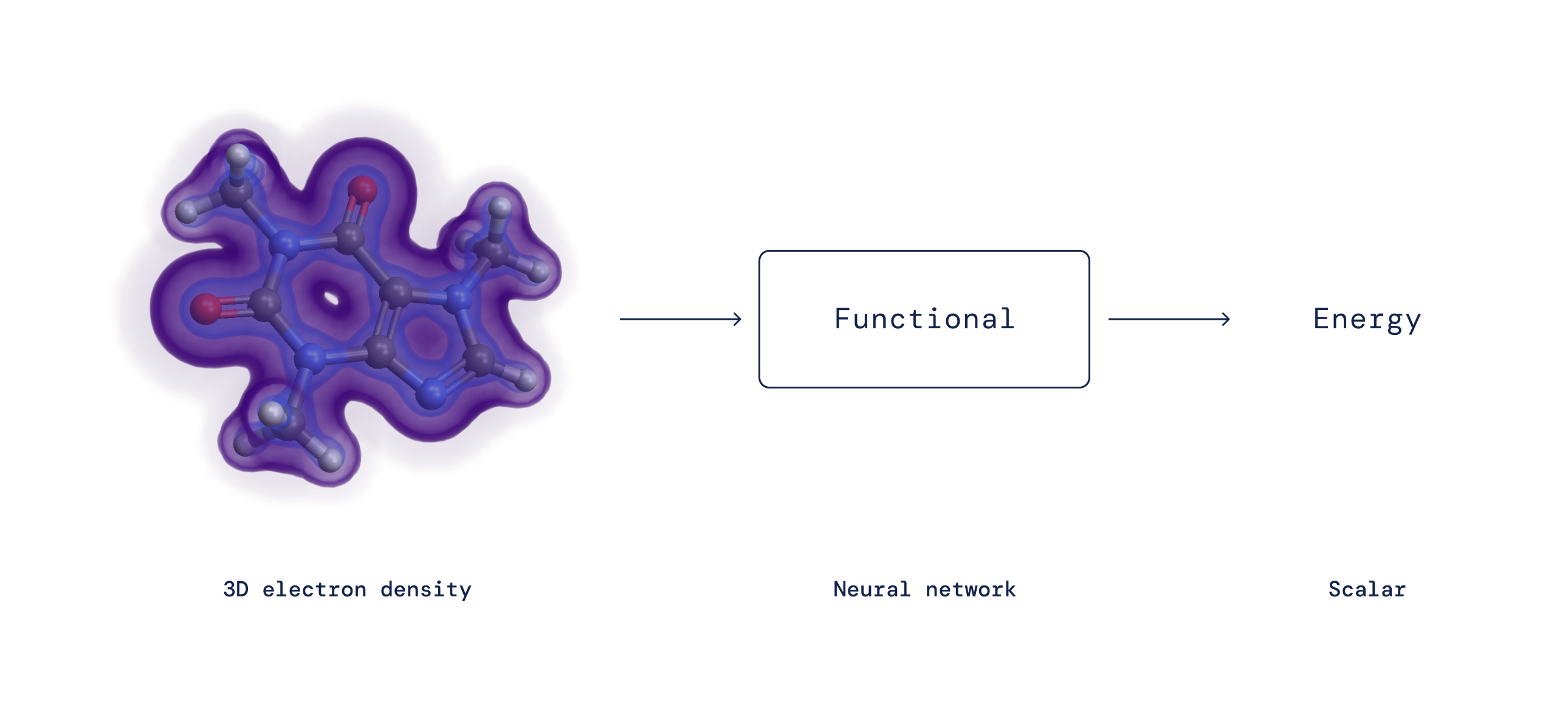
Почти столетие назад Эрвин Шредингер предложил его знаменитое уравнение управляющие поведением квантовомеханических частиц. Применение этого уравнения к электронам в молекулах является сложной задачей, поскольку все электроны отталкиваются друг от друга. Казалось бы, это требует отслеживания вероятности положения каждого электрона — чрезвычайно сложная задача даже для небольшого числа электронов. Один крупный прорыв произошел в 1960-х годах, когда Пьер Хоэнберг и Вальтер Кон поняли, что нет необходимости отслеживать каждый электрон по отдельности. Вместо этого, зная вероятность любой нахождения электрона в каждой позиции (т. е. электронной плотности) достаточно для точного расчета всех взаимодействий. Кон получил Нобелевская премия по химии после доказательства этого, тем самым основав функциональную теорию плотности (DFT).
Хотя DFT доказывает, что отображение существует, более 50 лет точная природа этого отображения между электронной плотностью и энергией взаимодействия — так называемый функционал плотности — оставалась неизвестной и должна быть аппроксимирована. Несмотря на то, что DFT по своей сути включает в себя уровень приближения, это единственный практический метод изучения того, как и почему материя ведет себя определенным образом на микроскопическом уровне, и поэтому он стал одним из наиболее широко используемых методов во всей науке. За прошедшие годы исследователи предложили множество приближений к точному функционалу с разным уровнем точности. Несмотря на свою популярность, все эти аппроксимации страдают от систематических ошибок, поскольку не могут уловить некоторые важные математические свойства точного функционала.
Выражая функционал в виде нейронной сети и включая эти точные свойства в обучающие данные, мы изучаем функционалы без важных систематических ошибок, что приводит к лучшему описанию широкого класса химических реакций.
Мы специально решаем две давние проблемы с традиционными функционалами:
- Ошибка делокализации: В расчете DFT функционал определяет плотность заряда молекулы, находя конфигурацию электронов, которая минимизирует энергию. Таким образом, ошибки в функционале могут привести к ошибкам в расчете электронной плотности. Большинство существующих приближений функционала плотности предпочитают электронные плотности, которые нереалистично распределены по нескольким атомам или молекулам, а не правильно локализованы вокруг одной молекулы или атома (см. рис. 2).
- Нарушение спиновой симметрии: при описании разрыва химических связей существующие функционалы имеют тенденцию нереалистично предпочитать конфигурации, в которых нарушена фундаментальная симметрия, известная как спиновая симметрия. Поскольку симметрии играют жизненно важную роль в нашем понимании физики и химии, это искусственное нарушение симметрии выявляет серьезный недостаток существующих функционалов.
В принципе, любой химико-физический процесс, связанный с перемещением заряда, может страдать от ошибки делокализации, а любой процесс, включающий разрыв связей, может страдать от нарушения спиновой симметрии. Перемещение заряда и разрыв связей являются ключевыми для многих важных технологических приложений, но эти проблемы также могут привести к массовой качественной несостоятельности функционалов для описания простейших молекул, таких как водород. Поскольку DFT является такой важной технологией, важно разработать функционалы, которые правильно понимают эту простую химию, прежде чем просить их объяснить гораздо более сложные молекулярные взаимодействия, такие как те, которые могут происходить в батарее или солнечном элементе.
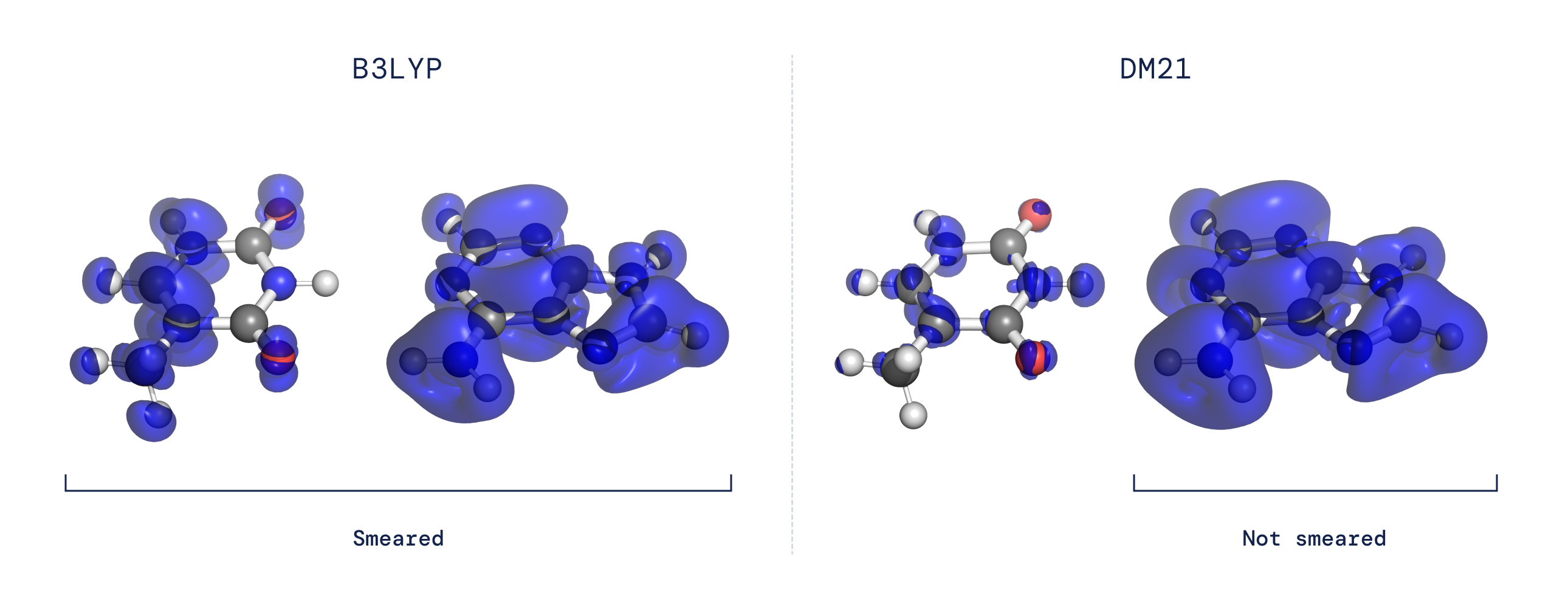
Обе эти давние проблемы связаны с тем, как ведут себя функционалы при работе с системой, демонстрирующей «дробный электронный характер». Используя нейронную сеть для представления функционала и адаптируя наш обучающий набор данных для захвата поведения дробных электронов, ожидаемого для точного функционала, мы обнаружили, что можем решить проблемы делокализации и нарушения спиновой симметрии. Наш функционал также показал себя очень точным в широких крупномасштабных тестах, предполагая, что этот подход, основанный на данных, может охватить аспекты точного функционала, которые до сих пор были неуловимыми.
В течение многих лет компьютерное моделирование играло центральную роль в современной инженерии, позволяя давать надежные ответы на такие вопросы, как «удержится ли этот мост?» на «вылетит ли эта ракета в космос?» По мере того, как технологии все чаще обращаются к квантовому масштабу для изучения вопросов о материалах, лекарствах и катализаторах, включая те, которые мы никогда не видели и даже не могли себе представить, глубокое обучение обещает точно моделировать материю на этом квантово-механическом уровне.